报错时考虑的重要命令:dos2unix
POTCAR中常用参数说明:
- VRHFIN 用来看元素的价电子排布,如果你元素周期表倒背如流,可以忽略这个参数;
- LEXCH 表示这个POTCAR对应的是GGA-PBE泛函;如果INCAR中不设定泛函,则默认通过这个参数来设定
- TITEL 就不用说了,指的是哪个元素,以及POTCAR发布的时间;
- ZVAL 指的是实际上POTCAR中价电子的数目,尤其是做Bader电荷分析的时候,极其重要。
- ENMAX 代表默认的截断能。与INCAR中的ENCUT这个参数相关。
如果想把体系从Cartesian转化为Direct,我们可以算一个单点。因为CONTCAR结果为Direct坐标系
电子结构优化:电子步
几何结构优化:离子步
电子步(SCF):EDIFF<=>NELM
离子步(结构优化):EDIFFG<=>NSW
电子迭代自洽算法:DAV(Blocked Davidson algorithm)、RMM(residual minimization scheme)、CG(conjugate-gradient algorithm)等等,在ICNAR中通过ALGO调整
自旋极化参数设置(ISPIN=1,不打开自旋极化;=2打开):
参照下列情况考虑自旋极化:
- 单原子的计算
- O2分子(基态为三重态)
- 自由基相关的计算
- 含Fe,Co, Ni 的体系
- 要计算的体系具有磁性:顺磁,铁磁,反铁磁等,要打开自旋极化
- 当关注体系的电子性质时,且自己不知道加或者不加的时候,建议加上
改变晶胞对称性对于原子电子结构计算很重要,如果只用对称性参数(ISYM=0时,不考虑对称性)进行描述是不够的(只能起到有限的作用)
ISMEAR:
金属体系:
ISMEAR一般用ISMEAR=0 或者整数1,2即可。
SIGMA =0.1 足够
气相分子
ISMEAR=0
SIGMA=0.02参考值(要小一点)
EDIFF&EDIFFG:
分别对应电子步的收敛判据和离子步的收敛判据,其中EDIFFG取1e-5即可,EDIFFG小体系用力的判据,取-0.01即可(注意负号),大体系用能量作为判断依据(1e-4即可,电子步收敛精度*10)
IBRION:
进行分子优化时需要引入
一般来说,优化结构的时候有3个选择:
IBRION=3:你的初始结构很差的时候;
IBRION=2:共轭梯度算法,很可靠的一个选择,一般来说用它基本没什么问题。
IBRION=1:用于小范围内稳定结构的搜索
IBRION=5:振动频率计算,此时POTIM要使用更小的数值,EDIFF需要更严格的值,加上NFREE=2,表示在某方向正反方向运动要(注意:最好把NCORE注释掉,不然只能算一步!)
MAGMOM:
初始磁矩设置,简单的磁性体系,我们可以直接使用ISPIN=2, MAGMOM不必进行设置。不知道体系的磁矩是多少,可以根据原子所处的化学环境, 根据成键情况,大体推测有多少个未成对电子,然后将未成对电子数目*1.5(注意!前面是数目,后面是磁矩)
MAGMOM = 10*-2 ——有10个原子,每个原子的初始磁矩为 -2
POTIM:
计算离子间作用力时,控制优化步离子移动的大小。
应用前提是IBRION=2,如果初始结构较好则随意,初始结构较差需要设置小一点(POTIM=0.05可以作为参考值)
ENCUT:
1.3*EMAX即可
ISTART:
0 :自动生成波函数(轨道)
1 :读取WAVECAR中的轨道信息,读不到就自动生成,根据新的设定重新生成平面波基组。
2 :读取WAVECAR中的轨道信息,读不到就自动生成,使用旧的平面波基组。
NCORE:
大师兄经验:NCORE = 单个节点核数 / 2 的时候,运行最省时间,设置也最方便。上交超算用40/2=20。计算频率的时候注释掉!(不然只能算一步,原因不明)。(计算单点的时候也注释掉!不然会出问题)(慎重使用!建议不用!)
从chemspider获取PSOCAR的时候,注意给的坐标是笛卡尔(Cartesian)还是分数坐标(Direct),分数坐标各个元素均小于1。
原子固定操作:
- 元素数目之后加入select关键字S
- 通过对应原子坐标最后T/F的设置控制固定(F)与放松(T)
振动频率计算要点:

conventional cell和primitive cell的区别:
conventional cell是搞晶体材料的经常用的,其特点是所选的cell在视觉上有非常好的对称性,但里面会存在重复的原子。也就是说,它不是晶体最小的重复单元。(一般用conventional cell)
primitive cell是晶体最小的重复单元。
关于K点设置的经验:
K * a = 45左右之间完全可以满足要求
LREAL:
确定投影在实空间求值(Auto等)还是在倒易空间(.FALSE.)求值
我们建议对包含20个以上原子的系统使用实空间投影方案。我们还建议只使用LREAL=Auto(对于版本VASP.4.4和更新的版本)和LREAL=On(对于所有其他版本)…通常使用LREAL=Auto可以获得最佳性能,但如果性能不是那么重要,也可以使用LREAL=.TRUE。
计算时要根据原子数的多少,以及后续的计算选择LREAL的值。不能拿LREAL=.FALSE.和LREAL=ON/.TRUE.计算的结果进行能量比较。
NELM:
设置电子SC(自一致性)步骤的最大数量(默认60)。通常,不需要更改默认值:如果自一致性循环在60个步骤内没有收敛,它可能根本不会收敛。在这种情况下,您应该重新考虑标记IALGO或ALGO、LSUBROT和混合参数。
ALGO:
指定电子最小化算法。或选择GW计算的类型。计算不收敛的时候多尝试修改这个参数。
ISIF:
是否计算应力张量。常用ISIF=2/3,使用时候需要增加ENCUT来减少误差【针对晶格常数!】。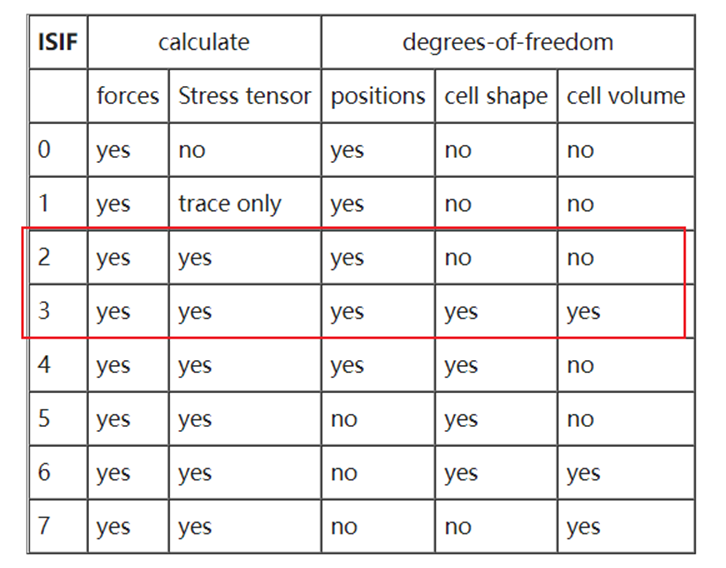
一旦我们计算完成晶格常数的计算后,可以在此结果基础上,统一使用其他的ENCUT值进行计算。也就是只有确定晶格常数的这一步是个例外,其他情况还是用原来的值。使用ISIF = 3 时,你脑子第一反应就是疯狂地增加ENCUT,600不够,上700, 700感觉不够,上800,在计算承受的范围之内,越大越好! 因此ISIF = 3 和 ENCUT两个参数必须同时出现。
晶胞优化的时候,一般用ISIF=3;
slab 模型优化的时候,我们用ISIF =2 ,也就是VASP的默认值,不用设置。
Slab模型说明:
Slab模型有两种,一种是上下表面对称的,一种是非对称的。对称性结构往往需要很多层,体系较大。 非对称的结构体系较小,但存在偶极矩的影响,要注意加LDIPOL 和IDIPOL这两个参数来消除:
块体材料计算时,为了后续计算其他性质,常常要求整个计算中使用相同的ENCUT,ENAUG, PREC, LREAL, ROPT。
一般来说用Conventional cell, FCC的金属可以用Primitive Cell 但对于其他体系,通过Primitive Cell切出来表面模型有问题。
偶极矫正:LDIPOL = .TRUE. ; IDIPOL = 3 Slab模型一般都需要加上这个参数
优化模型的时候需要考虑固定底层原子!在真实的环境下,催化剂体相看做是不变的,只有表面的原子参与催化反应。
Slab固定说明:
VASP中固定原子需要在POSCAR中进行操作。有两个关键点:
1)在POSCAR的第7行后添加一行,改行内容为Selective Dynamics,VASP只认第一个字母,你可以直接在这一行只加 S或者s。也可以换成其他的S开头的单词,比如SB,Sexy BigBro , Super BigBro等等;
2)在原子坐标后面加上 F 或者 T 表示固定或者放开,因为坐标有xyz三个数值,因此我们需要三个F或者T。我们可以通过设置允许原子在某一方向上移动,而其他方向上固定。
F F F表示xyz全部固定;
T T T 表示xyz全部放开,
F F T 表示 xy方向固定,只允许原子在z方向上移动。
PREC:
确定计算的精度模式,常常与ENCUT兼容使用。PREC=High;ENCUT=1.3Emax;
计算频率的时候 PREC=Normal(vasp5.默认)
ICHARG:
ICHARG确定VASP如何构造初始电荷密度。
Default: ICHARG = 2 if ISTART=0
LCHARG&LWAVE:
LCHARG determines whether the charge densities (files CHGCAR and CHG) are written.
LWAVE determines whether the wavefunctions are written to the WAVECAR file at the end of a run.